 |
John B. PariseDistinguished Toll Professor Ph.D., James Cook University, 1981 Lecturer, University of Sydney, 1986-87 Visiting Scientist, E.I. DuPont, 1981-83, 1988-89 Faculty member at Stony Brook since 1989 |
Professor Parise is a mineralogist, solid-state chemist interested in materials synthesis. He directs the US-DOE Energy frontier Research Center - GENESIS: a net GENeration synthESIS Center, based at Stony Brook. The GENESIS EFRC focuses on synthesis science. The broad goal is understanding of how specific reaction pathways and products can be targeted by tuning reaction parameters. The solutions team focuses on using in situ studies to comprehensively map reaction space in a high-throughput manner at lab-based and synchrotron sources. More information is available at https://www.stonybrook.edu/genesis. Parise also directs the Joint Photon Sciences Institute (https://you.stonybrook.edu/jpsi/news-and-events/ ) which is a hub for developing and applying X-ray methods, and supporting researchers using the nearby NSLS-II synchrotron and the upcoming Lab-EXAFS User Facility
The Parise group maintains an eclectic mix of projects, tied together by one guiding principal: We care about making materials, where the atoms are, and where atoms end up after changes in environmental conditions. No matter what the particular composition of the condensed matter its functionality is dependent on where the atoms are and where they end up after they respond to changes in P, T, Eh, pH, etc) - think graphite - diamond, Ice - water, DNA to remind yourselves of the fundamental need to determine where the atoms are under the operating conditions of interest.
The group places a strong emphasis on synthesis and students make the samples they later characterize.
The structure of liquids, nano-crystalline and amorphous materials:
Increasingly, solutions proposed to a number of technological challenges in the energy sector involve the synthesis and/or use of disordered materials, including liquids, melts, nano- and glassy materials, at extreme conditions. Examples include materials under high radiation fluxes advanced nano-materials for energy storage and conversion, clathrates and other materials in contact with sequestered carbon - supercritical CO2 in reservoirs or the deep ocean, for example, and high energy density materials. Understanding how the atomic arrangements in this class of materials respond to changes in pressure (p), temperature (T) gas loading and high chemical gradients is therefore fundamental to 1) understanding the behavior of these materials under their operating conditions, and 2) deriving general principles applicable to such classes of materials that 3) can be applied to vary conditions of manufacture that "steer" toward the desired product. Addressing our energy-related problems will therefore involve learning how to vary reaction pathways, including those at extreme conditions, in order to obtain precise information on changes in structural arrangements in situ. We combine development of sample cells for in situ x-ray and neutron (XN) scattering of key classes of materials under their operating conditions, with innovative data analysis and modeling in order to characterize materials with intrinsic disorder, such as are encountered in nano-, melt, glassy and gas adduct materials. We do this in collaboration with partners in industry at the Advanced Photon Source (APS) at Argonne National Laboratory (ANL) in Chicago, at the National Synchrotron Light Source-II (NSLS-II) at Brookhaven National Laboratory (BNL) some30 minutes from the Stony Brook University (SBU) campus, and at the Spallation Neutron Source (SNS) at Oak Ridge National Laboratory (ORNL) in Tennessee.
The "structure" of UO2 in solid and molten forms:
|
A recent study of UO2 melt is illustrative of the philosophy of choosing difficult first order problems and using scattering and analysis to provide fundamental insights into behavior and properties. The motivation for this study is the extensive use of uranium dioxide (UO2) as the major fuel component for most nuclear power reactors in use today (even mixed oxide (MOX) fuel is typically 90wt% UO2). A key safety concern in nuclear accidents is the melting and subsequent leakage of fuel. However the very high melting temperature (3140K) has limited study of the liquid phase. Consequently, relatively little is known about the atomic structure of molten UO2, and to our knowledge no detailed diffraction measurements have been made. Whilst several empirical potential Molecular Dynamics (MD) models exist for UO2, these are often constructed from fitting to low temperature crystal phases. The temperature-range of applicability of these models is often limited, and in the absence of measurements their relevance to the liquid structure remains un-validated. In UO2,the extension of empirical MD models is also complicated by the pre-melting lambda transition: The liquid is likely to be more similar to the post transition solid, than to the lower-temperature solid, from which most MD potentials are derived; MD simulation is used to derive such important parameters as viscosity, and so validation of potentials using the liquid scattering function above the melting point is a primary measurement that will validate potentials. |
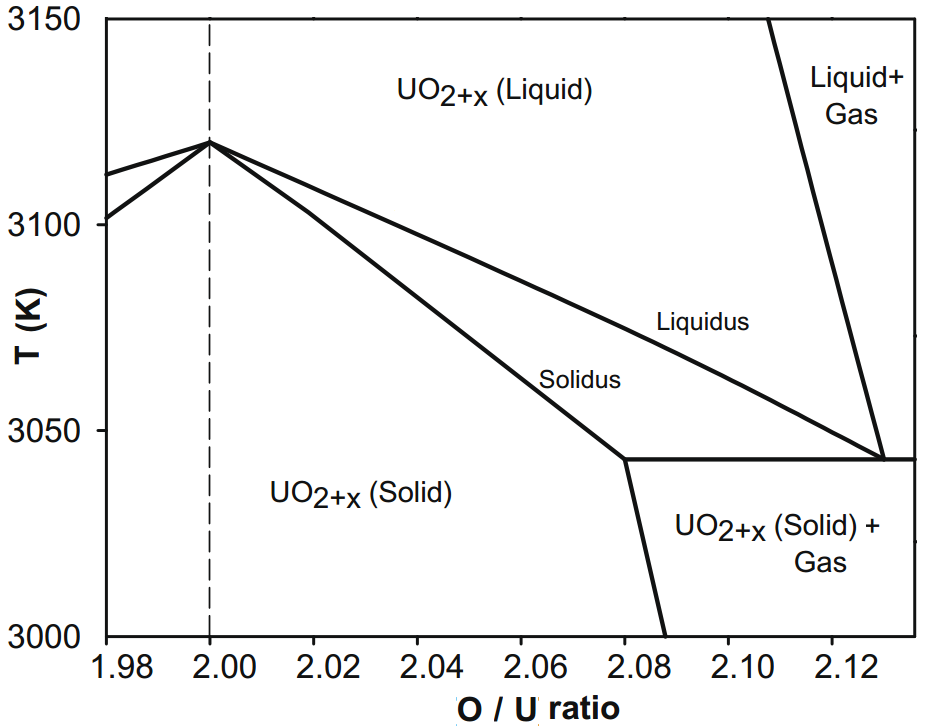 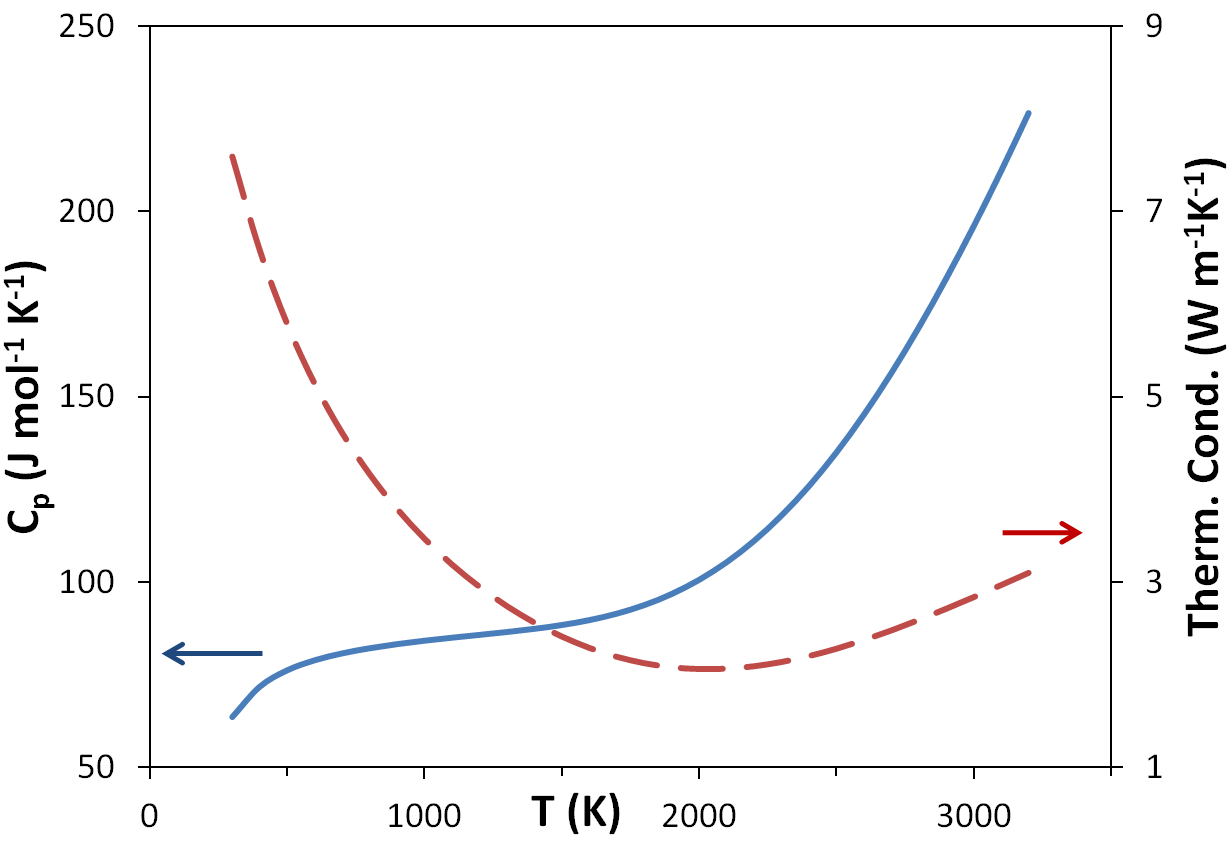 Top: Phase diagram around the melting point of UO2. Bottom: Heat capacity and conductivity of UO2 approaching the superionic lambda transition at 2670K |
Highlight articles referring to this work, published in Science (Skinner, L. B.; Benmore, C. J.; Weber, J. K. R.; Williamson, M. A.; Tamalonis, A.; Hebden, A.; Wiencek, T.; Alderman, O. L. G.; Guthrie, M.; Leibowitz, L.; Parise, J. B. Molten uranium dioxide structure and dynamics Science 2014, 346, 984-987) can be found here:
https://www.sciencenews.org/article/radioactive-fuel-turns-goo-during-nuclear-meltdown
|
We leveraged developments in high-powered laser heating and aerodynamic levitation detector corrections and experience with general rules recently derived for the behavior of oxide melts. The levitation technique involves the floating of a 3mm ball-shaped sample on an upwards gas stream. The absence of a solid contact surface allows the chemical purity of the very hot sample to be maintained. Also a low oxygen atmosphere will be needed to avoid 3(UO2) + O2 → U3O8 at 970 K. A short summary of results is provided in the highlights. The scientific program is underpinned by sound technical developments now that now allow us to study a variety of important disordered materials in situ, over a wide range of conditions, with unprecedented precision. Novel cell-design will be particularly important as operations begin at NSLS-II. |
 |

The first structure measurements of molten UO2 at 3000°C have been carried out recently at the APS. These experiments were enabled
by development of atmosphere controlled levitation and laser heating techniques. UO2 is the primary ingredient for nuclear fuel. Working with very hot liquids is technically
challenging. The new structure data will advance models used to predict properties
and behavior of UO2 at extreme conditions. Samples were levitated in high purity argon and heated to
3000 °C with a 400 W CO2 laser. The structure of the liquid was measured in-situ using
a 111 keV x-ray beam. Data were acquired using an area x-ray detector with fast data
acquisition. Analysis was performed using custom software developed as part of this
project
Local structural variation with oxygen fugacity in Fe2SiO4+xfayalitic iron silicate melts
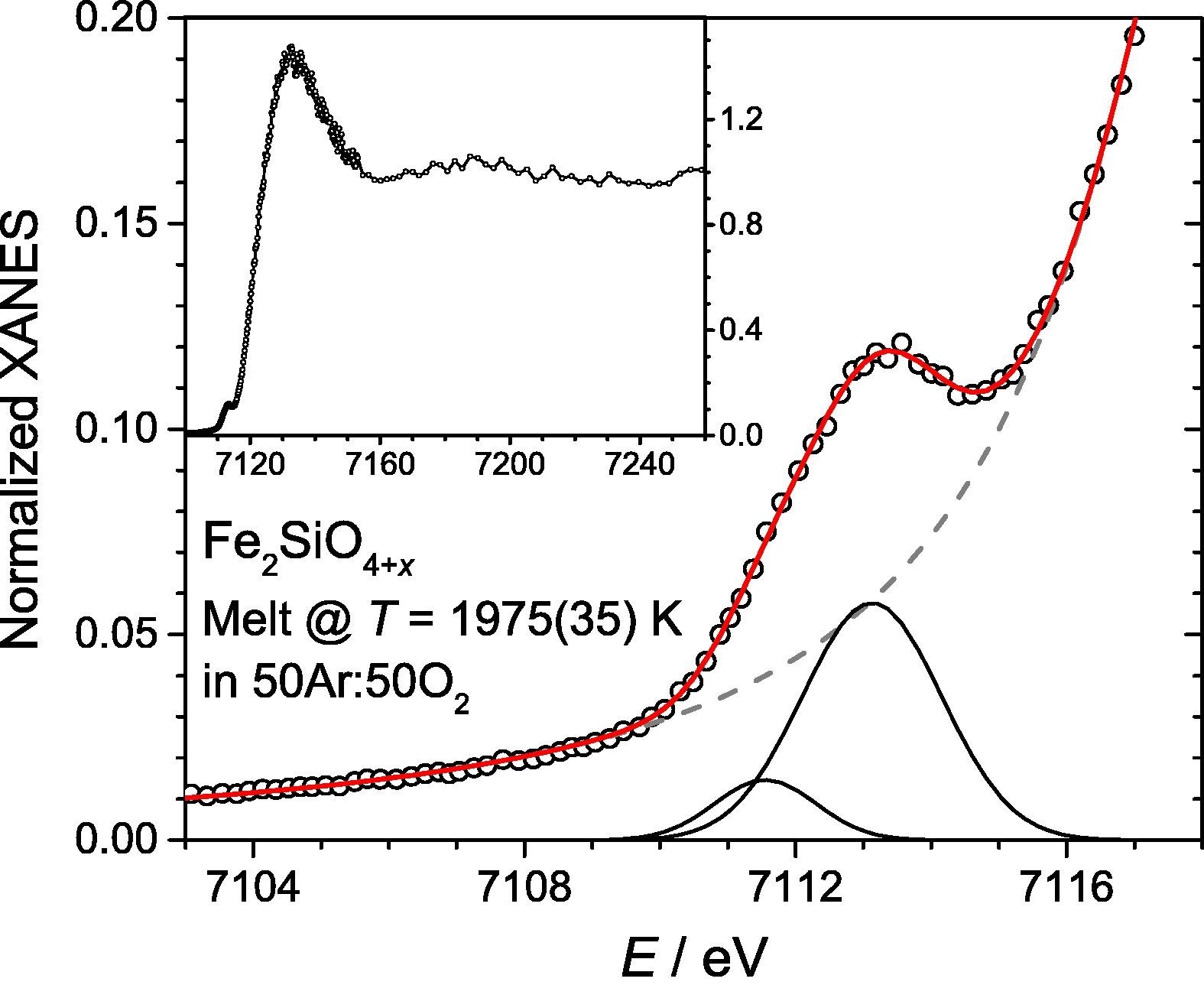
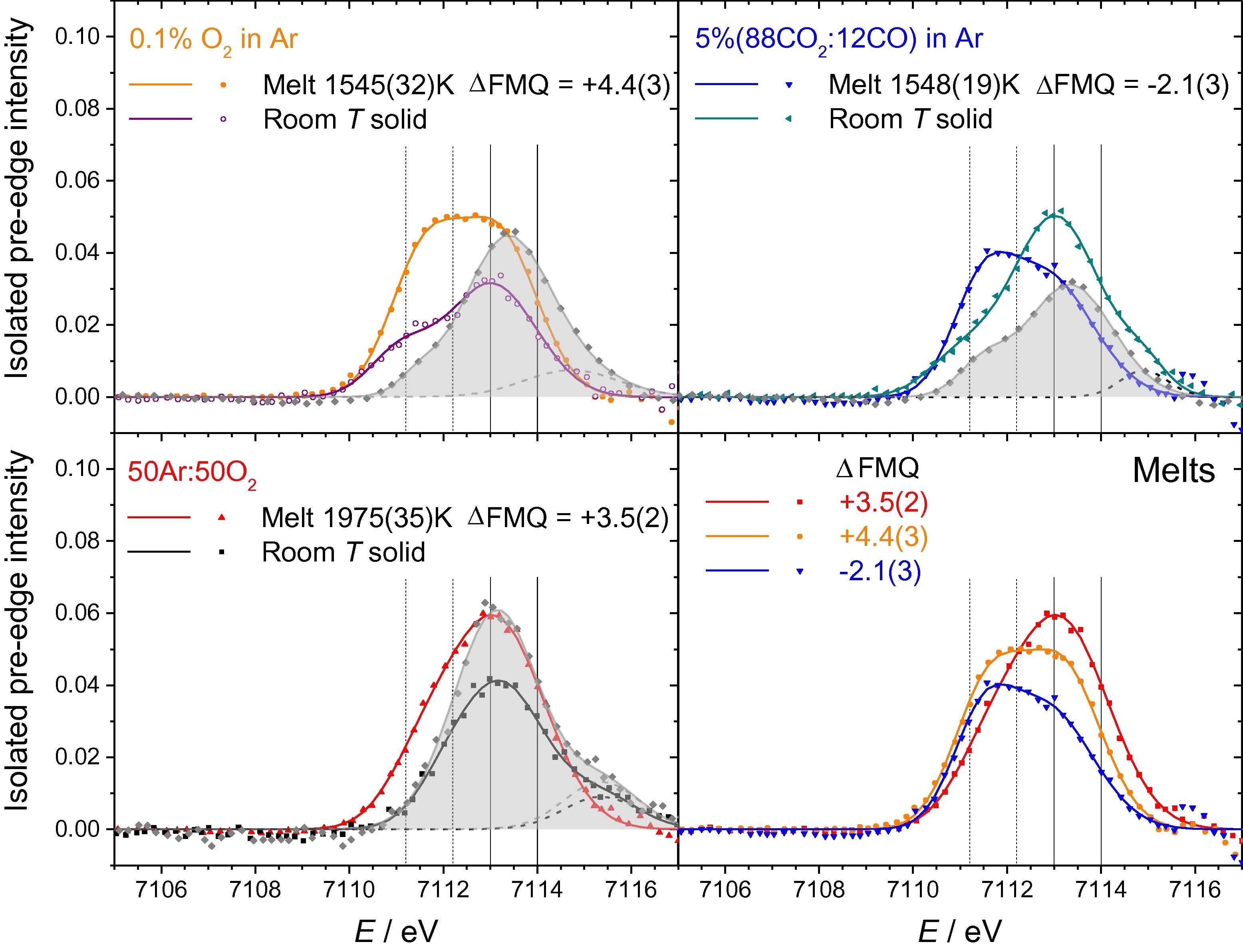
The structure of molten Fe2SiO4+x has been studied using both high-energy X-ray diffraction and Fe K-edge X-ray absorption near-edge structure (XANES) spectroscopy, combined with aerodynamic levitation and laser beam heating. A wide range of Fe3+ contents were accessed by varying the levitation and atmospheric gas composition. Diffraction measurements were made in the temperature (T) and oxygen partial pressure ranges 1624(21) < T < 2183(94) K (uncertainties in parentheses) and −5.6(3) < ΔFMQ < +2.8(5) log units (relative to the Fayalite-Magnetite-Quartz buffer). Iron K-edge XANES measurements covered the ranges 1557(33) < T < 1994(36) K and −2.1(3) < ΔFMQ < +4.4(3) log units. Fe3+ contents, x = Fe3+/ΣFe, estimated directly from the pre-edge peaks of the XANES spectra varied between 0.15(1) and 0.40(2). While these agree in some cases with semi-empirical models, notable discrepancies are discussed in the context of the redox kinetics and the limitations in both the models and in the calibrations used to derive oxidation state from XANES spectra. XANES pre-edge peak areas imply average Fe–O coordination numbers, nFeO, close to 5 for all Fe3+/ΣFe. Diffraction measurements yielded values of 4.4(2) < nFeO < 4.7(1). There is limited evidence for a linear trend nFeO(x) = 4.46(3) + 0.4(1)x. Asymmetric Fe–O bond length distributions peak at around 1.96 Å and have a shoulder arising from longer interatomic distances. Mean rFeO lie close to 2.06 Å, consistent with nFeOclose to 5. These observations suggest that Fe2+ is less efficient at stabilizing tetrahedral Fe3+ compared to large monovalent alkali cations.
Comparison of in-situ XANES estimates of Fe3+/ΣFe in the melts to those of the quenched solids obtained from XANES as well as Mössbauer spectroscopy indicate rapid oxidation during cooling, enabled by stirring of the melt by the levitation gas flow. As such, the oxidation state of hot komatiitic and other highly fluid melts may not be retained, even during rapid cooling, as it is for cooler basaltic and more silicic magmas
 |
 |
|
Left: Fe K-edge XANES spectrum for molten Fe2SiO4+x (1975(35) K) in an oxidizing atmosphere (50Ar:50O2). The measured pre-edge region is shown (open points) with bi-Lorentzian fit to the
main edge (dashed grey), two fitted Gaussian pre-edge peak components (solid black)
and the sum (solid red). Inset: XANES data over an extended energy range.
Right: Isolated pre-edge features of the Fe K-edge XANES spectra of molten and quenched
Fe2SiO4+x. Both experimental data (points) and fits (solid curves) are shown, with peaks assigned
to non-local transitions shown by broken curves. Each plot compares the melt to the
recovered quenched product measured as bulk bead and in powder transmission (shaded
area curves), except for the lower right plot which compares spectra for three melts
at different f(O2). See Table 2, Table 3. Vertical lines denote regions integrated over to extract the average Fe oxidation
state by the ratio method of Wilke et al. (2005). Note that the spectra for the solid (FMQ) beads may be affected by inhomogeneity and preferred orientation of crystallites, and also have larger uncertainties in their amplitudes owing to self-absorption
effects of surface roughness and shape irregularity not present for the molten (spherical) beads, or for the transmission
measurements.
|
|
Engineering frameworks that are tailored to be selective:
Zeolites and synthetic molecular sieves are useful materials for separations technologies. We are interested in not only how natural systems exchange, sequester and release ions, but also in how to apply the knowledge derived from basic science studies of natural systems to synthetic materials as well. The functionality of these as well as naturally occurring materials such as aluminosilicate zeolites, need to be understood in the context of their atomic structures. Separations applicable to the nuclear industry illustrate this point.
Capture of highly volatile radioactive iodine is a promising application of metal-organic frameworks (MOFs), thanks to their high porosity with flexible chemical architecture. Specifically, strong charge-transfer binding of iodine to the framework enables efficient and selective iodine uptake as well as its long-term storage. As such, precise knowledge of the electronic structure of iodine is essential for a detailed modeling of the iodine sorption process, which will allow for rational design of iodophilic MOFs in the future. Here we probe the electronic structure of iodine in MOFs at variable iodine...framework interaction by Raman and optical absorption spectroscopy at high pressure (P). The electronic structure of iodine in the straight channels of SBMOF-1 (Ca-sdb, sdb = 4,4 ' sulfonyldibenzoate) is modified irreversibly at P > 3.4 GPa by charge transfer, marking a polymerization of iodine molecules into a 1D polyiodide chain. In contrast, iodine in the sinusoidal channels of SBMOF-3 (Cd-sdb) retains its molecular (I-2 ) character up to at least 8.4 GPa. Such divergent high-pressure behavior of iodine in the MOFs with similar port size and chemistry illustrates adaptations of the electronic structure of iodine to channel topology and strength of the iodine...framework interaction, which can be used to tailor iodine-immobilizing MOFs.
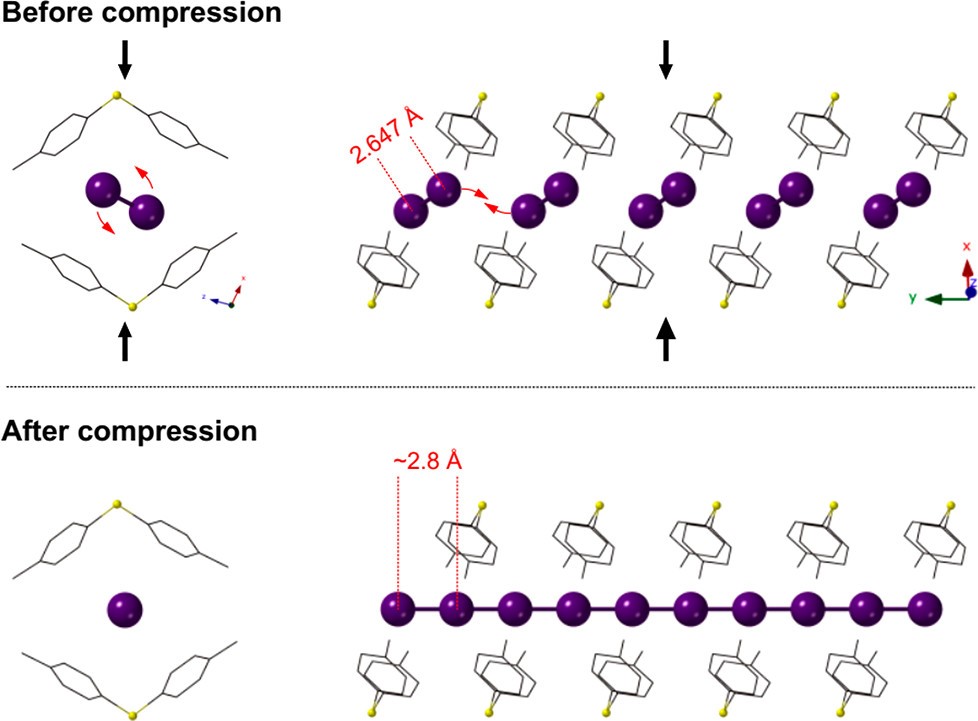 |
|
A spectroscopic model of the pressure-induced iodine polymerization in I@SBMOF-1.
Black arrows depict the main flexing direction of the I@SBMOF-1 unit cell.
|
Selected Publications
Solution/precipitation studies
[1] Beauvais, M. L.; Monserrate, B. A. S.; Mi, J. S.; Rossini, A. J.; Parise, J. B.; Chapman, K. W. Non-stoichiometry Governs the Pathway from Amorphous to Crystalline Calcium Carbonate. Journal of the American Chemical Society 2025, 147 (20), 17181-17189, Article. DOI: 10.1021/jacs.5c02800.
[2] Monserrate, B. A. S.; Beauvais, M. L.; Vornholt, S. M.; Chupas, P. J.; Parise, J. B.; Chapman, K. W. Real-Time Multiscale Imaging of Heterogeneous Multistage Reactions: Insights into Nanoscale TiO2 Synthesis. Journal of the American Chemical Society 2024, 146 (15), 10745-10752, Article. DOI: 10.1021/jacs.4c00749.
Liquids
[1] Skinner; Benmore; Weber; Du; Neuefeind; Tumber; Parise Low Cation Coordination in Oxide Melts Phys. Rev. Lett. 2014, 112, 157801
[2] Skinner; Benmore; Weber; Williamson; Tamalonis; Hebden; Wiencek; Alderman; Guthrie; Leibowitz; Parise Molten uranium dioxide structure and dynamics Science 2014, 346, 984.
[3] Alderman; Lazareva; Wilding; Benmore; Heald; Johnson; Johnson; Hah; Sendelbach; Tamalonis; Skinner; Parise; Weber Local structural variation with oxygen fugacity in Fe2SiO4+x fayalitic iron silicate melts 2017, 203, 15.
[4] Wilding; Wilson; Ribeiro; Benmore; Weber; Alderman; Tamalonis; Parise The structure of liquid alkali nitrates and nitrites 2017, 19, 21625.
[5] Wilson; Ribeiro; Wilding; Benmore; Weber; Alderman; Tamalonis; Parise Structure and Liquid Fragility in Sodium Carbonate J. Phys. Chem. A 2018, 122, 1071.
High Pressure Materials
[6] Woerner; Qian; Oganov; Stephens; Dharmagunawardhane; Sinclair; Parise Combined Theoretical and in Situ Scattering Strategies for Optimized Discovery and Recovery of High-Pressure Phases: A Case Study of the GaN-Nb2O5 System Inorg. Chem. 2016, 55, 3384.
[7] James; Esfahani; Woerner; Sinclair; Ehm; Oganov; Parise Theoretical and Experimental Investigations into Novel Oxynitride Discovery in the GaN-TiO2 System at High Pressure Crystals 2018, 8, 15.
[8] Dharmagunawardhane; James; Wu; Woerner; Palomino; Sinclair; Orlov; Parise Unexpected visible light driven photocatalytic activity without cocatalysts and sacrificial reagents from a (GaN)(1-x)(ZnO)(x) solid solution synthesized at high pressure over the entire composition range RSC Adv. 2018, 8, 8976.
Microporous Materials
[9] Banerjee; Simon; Plonka; Motkuri; Liu; Chen; Smit; Parise; Haranczyk; Thallapally Metal-organic framework with optimally selective xenon adsorption and separation Nat. Commun. 2016, 7.
[10] Banerjee; Wang; Plonka; Emge; Parise; Li Direct Structural Identification of Gas Induced Gate-Opening Coupled with Commensurate Adsorption in a Microporous Metal-Organic Framework 2016, 22, 11816.
[11] Banerjee; Chen; Lobanov; Plonka; Chan; Daly; Kim; Thallapally; Parise Iodine Adsorption in Metal Organic Frameworks in the Presence of Humidity ACS Appl. Mater. Interfaces 2018, 10, 10622.